Powered By



 Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Epilepsy is a condition that causes repeated seizures due to unusual electrical activity in the brain. Doctors may diagnose it using tests like an electroencephalogram (EEG) and magnetic resonance imaging (MRI).
There are many types of epilepsy, and each one can cause different symptoms. Knowing your type of epilepsy can help you and your doctor know what seizures to expect, how epilepsy may affect learning or development, and which treatments may help.
There are three major categories of seizures depending on where the seizures start. Some types of epilepsy are called syndromes, which means they have a pattern of signs and symptoms linked to a specific condition. The three categories of seizures are:
Read more about types of seizures.
Each type of epilepsy has its own features and pattern of brain activity. Some syndromes are considered benign, which means children may stop having seizures as they get older, while other types of epilepsy last for life.

Read on to learn more about 11 types of epilepsy.
About 60 percent of people with focal epilepsy have temporal lobe epilepsy (TLE), which causes seizures that begin in the brain’s temporal lobes. The temporal lobes are on the sides of the brain near the ears. They help process sound, language, and some memories linked to sound and vision.
About one-third of TLE or other cases of focal epilepsy do not respond to antiepileptic drugs (AEDs). Doctors may recommend epilepsy surgery to remove part of the brain or devices such as vagus nerve stimulation to help control seizures.
TLE usually develops between late adolescence and early adulthood, often after a head injury or febrile (fever-induced) seizure. Hormonal changes during the menstrual cycle can sometimes make focal seizures happen more often. This is called catamenial epilepsy.
Focal-onset seizures are most common for people with TLE, though some may experience prolonged seizures or, in rare circumstances, status epilepticus (repeated, long-lasting seizures).
Frontal lobe epilepsy is the second most common type of focal epilepsy. This type may be either inherited or caused by a structural problem such as a birth defect, an abnormal blood vessel, trauma, or scarring caused by infection. In about 50 percent of FLE cases, no cause is ever determined.
The frontal lobes are large areas at the front of the brain. They help regulate personality, behavior, and cognitive function (learning, thinking, and decision-making). FLE seizures frequently happen at night and can be mistakenly diagnosed as sleep disturbance or mental health problems.
Symptoms can vary a lot depending on which part of the lobe is affected. Frontal lobe epilepsy is typically treated with medication. But if AEDs don’t work, surgery or neuromodulation may help, including vagus nerve stimulation, responsible neurostimulation, or deep brain stimulation.
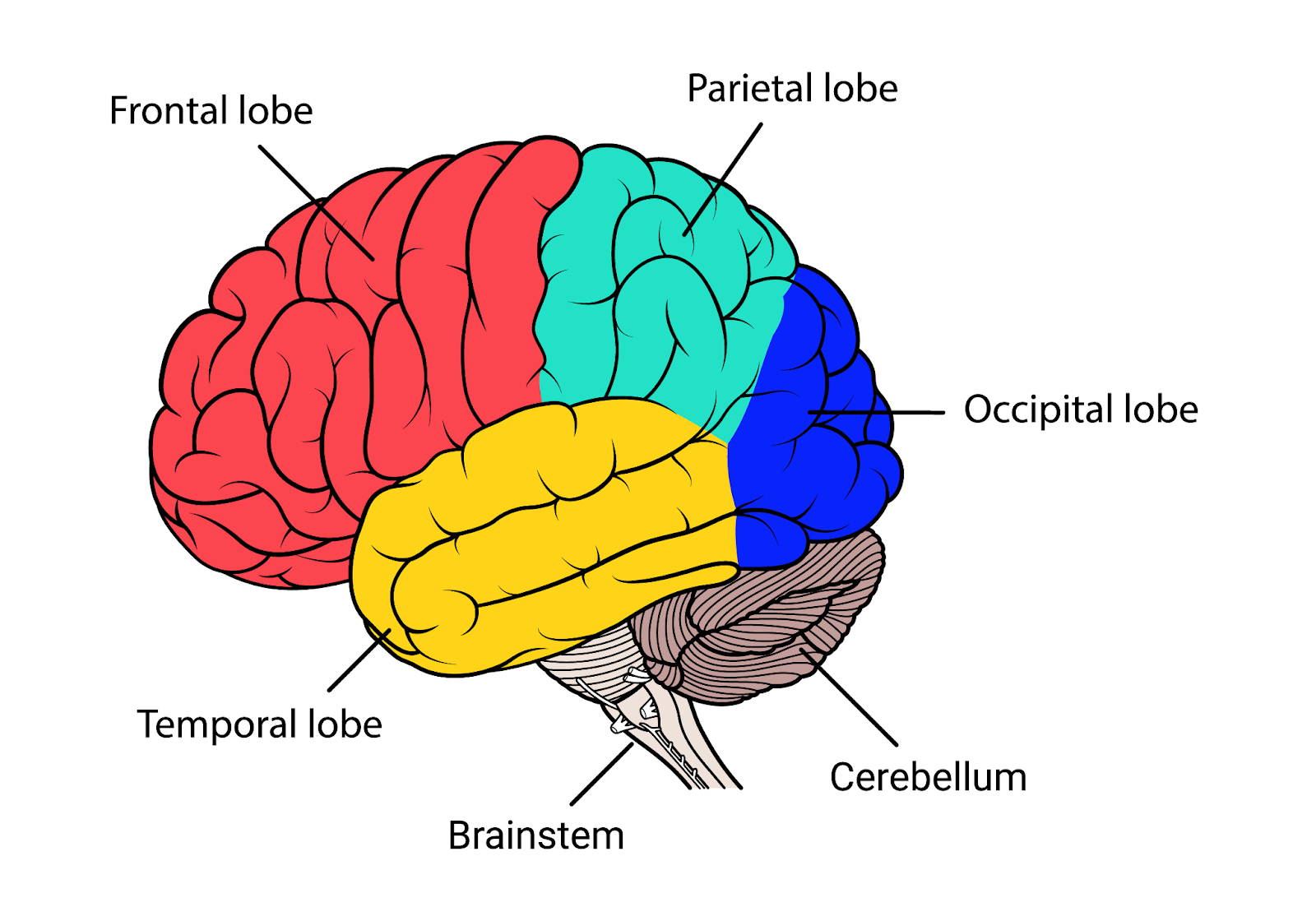
The occipital lobes are in the back of the brain and mainly process vision. Occipital lobe epilepsy is a rare type of focal epilepsy that affects sight. The cause of seizures that begin in the occipital lobe is often unknown but may be due to lesions in the occipital lobe from a brain injury, a birth defect, or genetic factors. AEDs are usually the first treatment option, though surgery may help if medications aren’t successful.
The parietal lobes are near the top of the brain and along the upper sides of the head. This area, called the association cortex, helps the brain make sense of sounds, images, and touch.
Focal epilepsy starting in the parietal lobe is much less common than TLE and FLE. Seizures that begin in the parietal lobes can cause sensory disturbances and sensations such as hallucinations, heat sensations, spatial distortion, difficulty understanding words, and numbness.
Some cases are caused by brain trauma, tumors, or stroke, but often the cause is unknown. AEDs are the first treatment option. If medication doesn’t work, surgery may be recommended.
Also called early childhood onset occipital epilepsy, Panayiotopoulos syndrome (PS) usually starts between 3 and 6 years of age, but it can develop later. Its cause is unknown. About 6 percent of children who have epilepsy have this syndrome.
Panayiotopoulos syndrome frequently stops two to three years after the first seizure. In rare cases, children with PS may develop other forms of epilepsy, such as juvenile myoclonic epilepsy (JME).
Children with PS typically have focal aware seizures that can spread to a generalized seizure. Seizures in PS last between one and 30 minutes, and more than half happen during sleep.
Typical symptoms include pale skin, a sick feeling, and vomiting. Some children also have tonic-clonic movements (stiffening and jerking of muscles).
Seizures that do not happen often may not require medication. However, if needed, AEDs usually work to control PS seizures. Neurologists often teach parents how to start rescue therapy and create an emergency plan for children with PS.
Benign rolandic epilepsy (BRE) — also known as benign epilepsy with centrotemporal spikes — usually begins around ages 6 to 8 years. According to the Epilepsy Foundation, boys are slightly more likely to have BRE than girls. BRE accounts for about 15 percent of all epilepsies in children.
Benign rolandic epilepsy is a type of focal epilepsy that can cause numbness, twitching, or tingling in the face or tongue. Seizures may make it hard to speak and may cause drooling. The child remains conscious during seizures, which are infrequent and occur mostly at night.
AEDs may be prescribed if the seizures happen during the day or disrupt sleep, but many children don’t need medication. Seizures stop by early adolescence in almost all children with BRE.
Childhood absence epilepsy (CAE) accounts for about 2 percent to 8 percent of childhood epilepsy cases. This generalized epilepsy typically begins between ages 3 and 11, most frequently between ages 5 and 8.
One-third of children with CAE have a family history of seizures, suggesting that this may be one of the types of genetic epilepsy. Siblings of children with CAE have a 10 percent chance of developing epilepsy.
Children with CAE experience absence seizures (formerly known as petit mal seizures). In this type of seizure, the child isn’t aware or responsive during seizures and may stare, blink, or roll their eyes upward. You may notice them making a chewing motion or other repetitive movements.
Seizures usually last less than 15 seconds, after which the child immediately returns to normal. The child usually isn’t aware that they had a seizure. Seizures may be infrequent or happen many times a day.
Some children with frequent CAE seizures have concentration and memory problems before seizures start, but most children with this type of epilepsy have normal development.
At least two-thirds of children with childhood absence epilepsy respond to treatment, and their seizures will end by midadolescence. If AEDs aren’t effective, the ketogenic diet may help.
However, 10 percent to 15 percent of children with CAE will develop other seizure types during adolescence.
For example, they may develop myoclonic seizures (“myoclonic” meaning short periods of jerking movements), generalized tonic-clonic seizures (formerly known as grand mal seizures), or both.
JME, also known as Janz syndrome, is another type of childhood epilepsy and is one of the most common generalized epilepsy syndromes. This syndrome tends to begin in the teen years.
Everyone with JME has myoclonic seizures, which cause sudden jerking in the arms and legs or throughout the body. Tonic-clonic seizures are also common with JME. Absence seizures happen in about one-third of people with JME and can be the first sign of JME.
Seizures often happen soon after waking and can be triggered by lack of sleep, too much alcohol, stress, or anxiety. Photosensitivity, which means seizures triggered by flashing or flickering light, affects more than one-third of people with JME.
Most cases of JME are treatable with AEDs, which are usually needed for life.
Juvenile absence epilepsy (JAE) is similar to childhood absence epilepsy, but it generally starts between ages 10 and 16 and is usually a lifelong condition. Between 1 percent and 2 percent of people with epilepsy have JAE. Although it’s rare to have a family history of seizures, the cause of JAE is thought to be genetic.
People with JAE usually have absence seizures that last 10 to 45 seconds. They tend to have fewer than one absence seizure per day, often during exercise. Around 80 percent of those with JAE will also have tonic-clonic seizures.
JAE also carries a higher risk of absence status epilepticus, a type of nonconvulsive status epilepticus and an emergency condition in which seizures can last for more than 30 minutes.
Most children with JAE develop normally, although they may have learning difficulties if they have frequent seizures. Sometimes, concentration and memory problems arise before seizures start. These issues often improve with the use of AEDs, which work well to treat JAE and generally must be taken for life.
Lennox-Gastaut syndrome (LGS) is a rare and severe generalized epilepsy syndrome, occurring in 3 percent to 4 percent of children with epilepsy. According to MedlinePlus, it’s more common in males. LGS usually appears in early childhood and in infancy.
Children with LGS experience different types of seizures, particularly tonic-clonic seizures, atonic seizures (causing a loss of muscle tone, often with falling or drop attacks), and atypical absence seizures. Developmental and behavioral problems occur among two-thirds of children with LGS.
LGS is sometimes caused by other conditions, but often the cause is unknown. The condition is refractory (difficult to treat) because it is usually resistant to AEDs. A newer treatment, cannabidiol (Epidiolex), may provide more help than AEDs.
One report found that a ketogenic diet reduced seizures by 50 percent or more in people with LGS. Surgery and vagus nerve stimulation have also been shown to be effective in treating LGS, according to another study.
Progressive myoclonic epilepsies (PMEs) are a group of rare syndromes and types of epilepsy characterized by a combination of tonic-clonic and myoclonic seizures that worsen over time. The most common seizure disorders in this category include:
Progressive myoclonic epilepsies usually begin between ages 6 and 16, depending on the condition. The cause is often hereditary but may be unknown.
Seizures in progressive myoclonic epilepsies are hard to control. As the condition progresses, people with PME develop cognitive (thinking and memory) and motor (movement) disabilities. Medications may work at first but become less effective over months or years as the disease progresses.
Most types of epilepsy bring difficulties beyond the seizures themselves. Epilepsy can affect mood, disrupt sleep, affect social interactions, lead to thinking and memory problems, cause bone health issues, and, in some cases, increase the risk of early death.
If you or your child needs help coping with epilepsy, talk to a healthcare provider about treatment options and other resources that may help you manage some of the daily challenges that come with epilepsy.
Sometimes, epileptic seizures aren’t easy to notice. If you think you or a loved one might be having seizures without realizing it, talk to a neurologist as soon as possible.
On MyEpilepsyTeam, people share their experiences with epilepsy, get advice, and find support from others who understand.
What kind of epilepsy do you or your child live with? What are your challenges? Let others know in the comments below.
Get updates directly to your inbox.



How many different meds do you need to take to control all the seizures or can they all be controlled with one med.?.


 Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Continue with Facebook
Continue with Email
Become a member to get even more





 Join
Join


 Your Privacy Choices
Your Privacy Choices

This is a member-feature!
Sign up for free to view article comments.
Keep your head up one day at a time
We'd love to hear from you! Please share your name and email to post and read comments.
You'll also get the latest articles directly to your inbox.